
Rôle de la génétique
Le rhumatisme psoriasique est une maladie inflammatoire chronique, cliniquement très hétérogène. Des facteurs génétiques, environnementaux, immunologiques et microbiologiques aux interactions complexes participent à la physiopathologie (Figures 1 à 3).
Le terrain génétique du rhumatisme psoriasique est encore mal caractérisé. Les variants sont nombreux et leur rôle respectif, dans les études d’associations, entre le terrain du psoriasis cutané et celui du rhumatisme psoriasique, est parfois difficile à différencier. Mais on peut affirmer que le rhumatisme psoriasique est une maladie polygénétique très fortement héréditaire.
Le risque de manifestation dans la fratrie par rapport à celui dans la population générale est bien plus élevé si l’on prend les exemples du psoriasis et de la polyarthrite rhumatoïde.
La présence de certains haplotypes serait plus fortement associée au rhumatisme psoriasique et à certains de ses phénotypes cliniques qu’au psoriasis.
Des analyses d’association à l’échelle du génome ont montré que certains polymorphismes dans le gène codant pour le récepteur de l’IL-23 (IL-23R), ainsi que des variantes dans l’expression et la signalisation du facteur nucléaire κB (NF-κB), étaient associés au rhumatisme psoriasique. De plus, des études d’association ont identifié d’autres allèles de risque tels l’IL-12A, l’IL-12B, l’IL-23R et les gènes qui régu- lent le NF-κB chez les patients atteints de rhumatisme psoriasique et psoriasis.
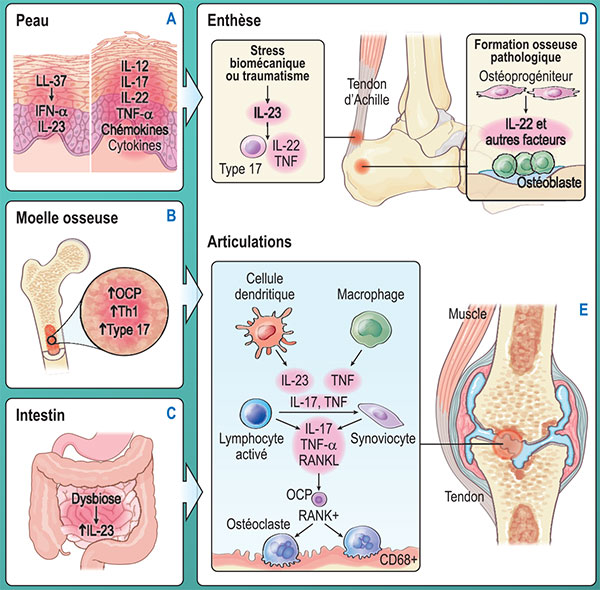
Figure 1. Voie de signalisation physiopathogénique dans le rhumatisme psoriasique (adapté de Ritchlin CT et al. N Engl J Med 2017 ; 376 : 957-70).
Légende de la figure1. L’enchaînement des événements potentiellement responsables de la survenue du rhumatisme psoriasique est décrit dans la figure. Des interactions entre des facteurs environnementaux et des facteurs génétiques sont à l’origine d’une réponse inflammatoire se développant à des sites multiples. Dans la plaque cutanée se formant, l’ADN libéré par les kératinocytes stressés se lie au peptide antibactérien LL-37 et stimule la libération d’interféron a par les cellules dendritiques plasmacytoïdes, les cellules dendritiques dermiques activées migrent vers les ganglions lymphatiques de drainage et déclenchent la différenciation des cellules T auxiliaires Th1 et Th17. Les cellulesTh1 et Th17 se réfugient dans le derme où elles libèrent des interleukines IL-12, IL-17 et IL-22, le TNFa et d’autres cytokines et chimiokines.
La libération de ces cytokines dans le derme favorise la prolifération des kératinocytes qui à leur tour libèrent des cytokines agissant de manière paracrine sur les cellules du derme (A).
L’expansion des cellules Th1 et Th17, d’autres cellules de type 17 et des précurseurs d’ostéoclastes (OCP) peut avoir lieu dans la moelle osseuse (B).
Une dysbiose microbienne peut déclencher une inflammation dans l’iléocolon et être à l’origine de la libération d’IL-23 et de cellules de type 17 (C).
Dans l’enthèse, la libération d’IL-23en réponse à un stress biomécanique ou à un traumatisme au niveau du site d’insertion du tendon, active les cellules de type 17 et d’autres cytokines, incluant l’IL-22 et le TNFa, qui peuvent être à l’origine d’inflammation, d’érosion osseuse et de formation osseuse pathologique. En réponse à l’IL-22 et à d’autres voies de signalisation, les cellules mésenchymateuses se différencient en ostéoblastes formant des enthésophytes au contact des enthèses et des articulations périphériques et des syndesmophytes au rachis (D).
À partir des enthèses ou de la circulation sanguine, les cellules de type 17, les OCP et les cellules dendritiques atteignent l’articulation adjacente. L’expression accrue du RANKL par les synoviocytes de la muqueuse associée à l’augmentation des taux d’IL-17, de TNFa et de RANKL exprimés par les cellules infiltrées, entraîne la différenciation des OCP en ostéoclastes matures,avec synovite et résorption osseuse(E).
Avec le soutien institutionnel des laboratoires