
- 1. L’évaluation préthérapeutique des hémopathies
- 2. Les anémies
- 3. Les leucémies aiguës
- 4. La leucémie lymphoïde chronique
- 5. Les lymphomes malins non hodgkiniens
- 6. Le lymphome hodgkinien
- 7. Les syndromes myélodysplasiques
- 8. Les syndromes myéloprolifératifs [1]
- 9. La leucémie myéloïde chronique
- 10. Le myélome multiple
10. Le myélome multiple
Le myélome multiple correspond à une prolifération médullaire maligne de plasmocytes pouvant sécréter une immunoglobuline (Ig) monoclonale ainsi qu’un facteur d’activation des ostéoclastes (cellules responsables de la résorption du tissu osseux par ostéolyse) [11].
Diagnostic/signes cliniques[1]
Les circonstances de découverte relèvent souvent de l’apparition des signes cliniques suivants :
- altération de l’état général, perte de poids ;
- signes osseux (douleurs, fractures pathologiques, tuméfactions osseuses) ;
- infections répétées et graves ;
- syndrome anémique ;
- syndrome d’hyperviscosité ;
- complication neurologique.
Le diagnostic se confirme principalement sur deux examens : le myélogramme, qui retrouve une infiltration plasmocytaire, et l’électrophorèse des protéines.
Des examens complémentaires sont nécessaires pour identifier une atteinte osseuse, une inhibition de l’immunité humorale, une atteinte rénale ou un syndrome d’hyperviscosité plasmatique.
Classification [1]
Les différentes formes cliniques
- Myélomes IgG
- Myélomes IgA
- Myélome à IgD
- Myélome IgE ou IgM (très rares)
- Myélome à chaînes légères
- Myélome non sécrétant
- Plasmocytome solitaire
- Leucémie à plasmocytes
Critères SLiM-CRAB
Les critères SLiM-CRAB déterminent l’intention de traitement. Les critères CRAB [41] définissent la présence ou non de symptômes cliniques :
- C: hypercalcémie ;
- R: insuffisance rénale ;
- A: anémie ;
- B: atteinte osseuse (bone) définie par pet-scan ou IRM.
Les critères SLiM, représentant des marqueurs de malignité, reflètent une maladie d’évolution rapide :
- S (Sixty) : Plasmocytose médullaire 6 60 % ;
- Li (Light chain): Ratio des CLL (chaînes légères libres) sériques impliquées/CLL non impliquées > 100 ;
- M (MRI): Plus d’1 lésion focale à l’IRM [41].
Classification de Durie et Salmon
(selon la masse tumorale) [Tableau X] [42]
[Tableau X] Classification du myélome multipleselon Durie et Salmon (d’après[42]).
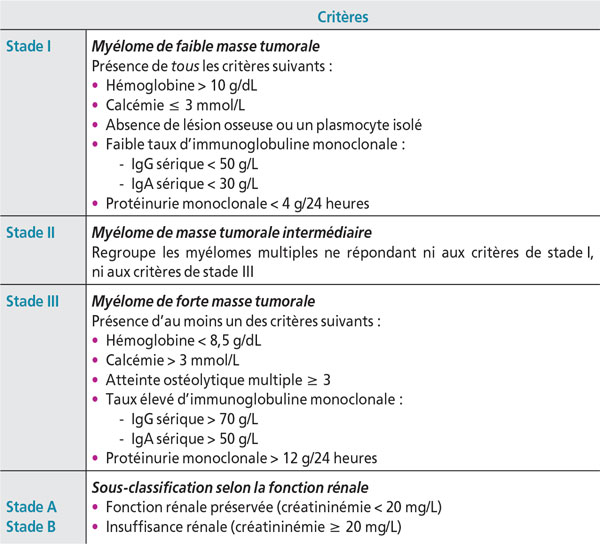
La classification de Salmon et Durie est de moins en moins utilisée. La découverte de plusieurs anomalies cytogénétiques de mauvais pronostic [délétion du bras court du chromosome 17 (del17p), translocation t(4;14)] vient compléter ces scores et va jouer un rôle important dans cette pathologie.
Complications [1]
• Osseuse
- Augmentation de l’ostéolyse (fracture, tassement)
- Crise d’hypercalcémie aiguë (asthénie, désorientation, syndrome polyurie-polydipsique, troubles cardiaques, possible sub-occlusion)
• Rénale
- Insuffisance rénale aiguë oligo-anurique
- Insuffisance rénale chronique avec diurèse conservée
- Syndrome néphrotique chronique
• Neurologique : compression médullaire ou radiculaire, poly- neuropathie périphérique
• Infectieuse : pneumopathie, infections urinaires
• Syndrome d’hyperviscosité : céphalées, vertiges,syndrome hémorragique, paresthésie, somnolence, flou visuel
• Amylose : signes cutanés, rénaux et cardiaques
Pronostic [1]
La stratégie thérapeutique est largement conditionnée par l’âge et la présence de comorbidités au moment du diagnostic. Le pronostic repose sur plusieurs paramètres comme le profil des anomalies cytogénétiques, le score R-ISS et ses variantes, l’âge et la fragilité du patient. Le score R-ISS (pour Revised-International stagingsystem) de l’IMWG [1] se base sur le taux de bêta- 2-microglobuline, l’albuminémie mais aussi sur le taux de LDH (lactate déhydrogénase) et la présence d’anomalies cytogénétiques incluant la translocation t(4;14) et/ou t(4;16) et/ou la délétion del(17p). La combinaison de ces marqueurs pronostiques permet également de stratifier les patients en trois groupes selon l’agressivité de la maladie[11].
Stratégies thérapeutiques [1, 11]
La décision du traitement repose sur les critères CRAB et, depuis la mise à jour des critères de l’International Myeloma Working Group (IMWG) en 2014, sur d’autres critères, SLiM, témoins d’une progression éminente comme le taux de plasmocytes médullaires supérieur à 60 %, le ratio kappa/lambda de chaînes légères/libres supérieur ou égal à 100 ou la présence de plus d’une lésion focale à l’IRM [41].
Polychimiothérapie
Le myélome multiple est traité par des associations de plusieurs médicaments dans le but d’obtenir la meilleure réponse possible en réduisant le clone tumoral. Les associations recommandées à l’heure actuelle comprennent 2 ou 3 voire 4 médicaments.
- Agents alkylants (chimiothérapies)
- Corticoïdes
- Inhibiteurs du protéasome (IP)
- Immunomodulateurs (IMiDs)
- Médicaments d’immunothérapie
Il s’agit ensuite, selon l’âge du patient et de son état de santé général (présence ou non de comorbidités), d’intensifier le traitement par une autogreffe de cellules souches hématopoïétiques.
Thérapies ciblées
Les inhibiteurs du protéasome
Ils altèrent les mécanismes de prolifération et de survie des cellules myélomateuses.
Les IMIDs
Les IMIDs sont des dérivés de l’acide glutamique, ils agissent sur les interactions entre les cellules tumorales et stromales, réduisent la migration métastatique et inhibent l’action de certains gènes impliqués dans le processus inflammatoire et immun du myélome.
Médicaments d’immunothérapie
Les marqueurs de biologie moléculaire présents à la surface des cellules tumorales représentent des cibles et ont permis de développer des médicaments d’immunothérapie, comme par exemple des anticorps qui favorisent entre autres l’action des cellules immunitaires contre les cellules tumorales. Ces médicaments peuventêtre combinés à de la chimiothérapie.
La multiple variété de ces traitements permet de changer de molécules à la rechute ou en cas d’évolution. Le myélome multiple devient une maladie chronique dont l’espérance de vie des personnes atteintes augmente [43].
Pour les patients ne présentant pas un des critères définissant le myélome multiple (IMWG 2014), la prise en charge consiste en une surveillance clinico-biologique et radiologique.
Avec le soutien institutionnel des laboratoires Janssen
