
- Introduction
- Lymphocytes de type 1, type 2 et type 17
- Immunité et inflammation de type 1, type 2, type 17
- Cytokines, récepteurs des cytokines et voies de signalisation intracellulaire
- Physiopathologie de l’inflammation cutanée
- Phase de résolution de l’inflammation
- Chronicité, rechutes et sévérité des dermatoses inflammatoires chroniques
- Introduction
- Facteurs pathogéniques de la maladie
- Présence d’altérations de la peau non lésionnelle
- Pertes des mélanocytes épidermiques
- Intervention de l’immunité innée
- Intervention de l’immunité adaptative de type 1
- Voies de signalisation impliquées dans le vitiligo
- Intervention de réponses immunitaires autres que celle du type 1
Installation de l’inflammation et progression de la maladie
Dans une première phase, nous avons décrit comment les cellules de l’immunité étaient recrutées sur le lieu de l’inflammation, revenons sur les types inflammatoires prédominants dans les lésions de la maladie :
• les cytokines IL-23 et IL-22, abondamment produites par les cellules dendritiques, sont associées à une inflammation de type 17, avec la production d’IL-17 par les LT17 ;
• l’IL-12, abondante aussi, favorise une inflammation de type 1avec production d’IFNγ par les LT1. L’IFNγ favorise l’activation des macrophages et des cellules tissulaires ;
• des LT22 sont aussi présents dans les lésions de la maladie, bien que de façon limitée. De fait, la synthèse d’IL-22 est partiellement inhibée par l’IL-10. Cette faible quantité d’IL-22 est associée à des taux de protéines antimicrobiennes (AMP) insuffisants pour contrer la croissance microbienne et l’inflammation de la peau.
L’action de l’ensemble de ces protagonistes et de leurs agents effecteurs conduit à une dilatation plus importante encore du follicule jusqu’à rupture. Cette rupture se produit sous l’effet combiné de la dégradation de l’épithélium, de l’érosion de la membrane basale et des mutations génétiques dont sont affectées les cellules souches épithéliales. Pris d’assaut par les cellules immunitaires, le follicule se transforme en abcès ou nodule inflammatoire :
• les fibroblastes et les kératinocytes produisent des métalloprotéinases (MMP1, 9, 10), qui sont des enzymes de dégradation de la matrice extracellulaire, et amorcent ainsi la dégradation tissulaire ;
• sous l’impulsion de la lipocaline 2 et du G-CSF (granulocyte colony- stimulating factor), les granulocytes infiltrent en masse les tissus et participent à la formation de pus ;
• les cytokines produites par les LT activés et les kératinocytes contribuent à l’hyperplasie épithéliale par l’intermédiaire de l’IL-36. L’hyperplasie s’étend jusque dans l’espace internodulaire.
La dégradation de la matrice extracellulaire, l’accumulation de pus et la dissémination de cellules souches dans le derme aboutissent à la formation de sinus et de fistules[11] (Figure 13).
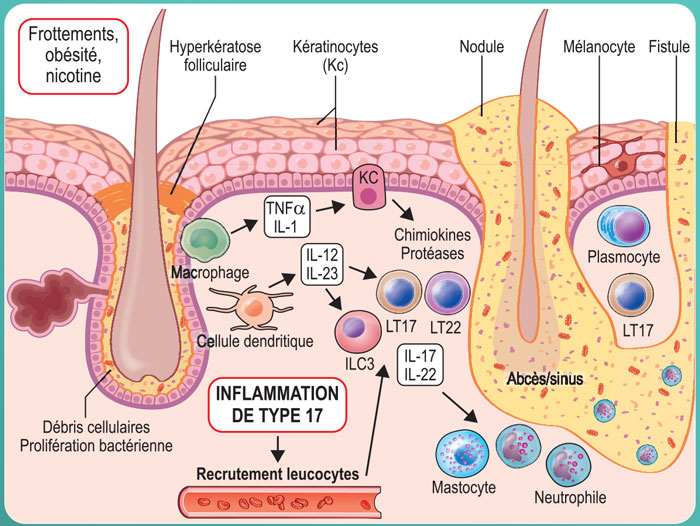
Figure 13. Mécanismes à l’origine de l’hydradénité suppurée. Progression de la maladie vers un stade avancé. Sous l’effet des cellules immunitaires présentes dans les lésions (lymphocytes T17, lymphocytes T22), l’inflammation se développe, l’invasion microbienne se répand, l’épithélium du follicule se dégrade, jusqu’à aboutir à une rupture du follicule, soumis à l’abcédation. Le nodule est pris d’assaut par les cellules immunitaires qui amorcent la dégradation tissulaire via la production de métalloprotéinases, la formation de pus par les granulocytes sous le contrôle de la lipocaline-2 et du G-CSF et l’hyperplasie épithéliale (via l’IL-36). L’ensemble de ces événements aboutit à la formation de sinus et de fistules.
Avec le soutien institutionnel de Pfizer
